
The US Food and Drug Administration (FDA) approved the first-ever diagnostic test to aid in the diagnosis of sexually transmitted Mycoplasma genitalium infection. The Aptima Assay from Hologic Inc, is the only FDA-approved test to detect this under-recognized but increasingly common sexually transmitted infection (STI).
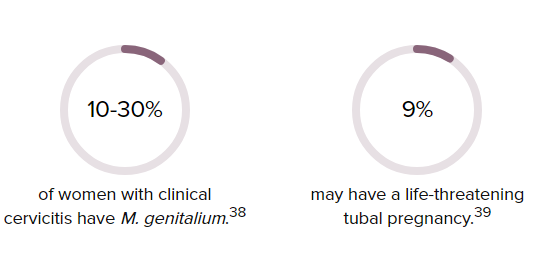
First discovered in the early 1980s, M. genitalium was listed as an emerging public health threat by the U.S. Centers for Disease Control and Prevention (CDC) in 2015. Currently, it is responsible for causing approximately 15 to 30 percent of persistent or recurrent urethritis cases in men in the United States and 10 to 30 percent of cervicitis cases in women.
This slow-growing pathogen is difficult to detect by traditional laboratory testing methods. In the absence of FDA approved diagnostic test, the STI is often misdiagnosed and treated with wrong antibiotics resulting in persisting infection and widespread transmission.

“Although Mycoplasma genitalium is typically more common than gonorrhea, there is very little public awareness of this rising sexually transmitted infection, which can cause serious and potentially devastating health problems,” said Damon Getman, Ph.D., senior principal research scientist and director of research at Hologic in a press release.
The Aptima Assay is a nucleic acid amplification test, which detects M. genitalium in urine samples and urethral, penile meatal, endocervical or vaginal swab collected in a clinical setting, such as a doctor’s office or clinic.
In the clinical study involving 11,774 samples, the Aptima test correctly identified M. gen. in approximately 90 percent of vaginal, male urethral, male urine and penile samples. It also correctly identified the presence of the pathogen in female urine and endocervical samples 77.8 percent and 81.5 percent of the time.
The test also has a high negative predictive value and correctly identified the negative samples 97.8 to 99.6 percent of the time. Vaginal swabs are the preferred samples to perform the test, but urine samples can be used as alternative sample types.
The assay is immediately available at clinical laboratories, physicians and healthcare providers can immediately order the tests by reaching out to the labs. Hologic further anticipates that most insurance plans will cover the testing for M. genitalium.
The Aptima Mycoplasma genitalium Assay was reviewed through the de novo premarket pathway, a regulatory pathway for some low- to moderate-risk devices of a new type. The FDA exercises special controls over the individual tests approved by the de novo premarket route to ensure the safety and effectiveness of the tests.
The newest M. genitalium assay joins the comprehensive list of men and women’s health assays by Hologic that include sexual and cervical health, and virology testing.







